Pathophysiological role of mitochondrial aconitase in unloading-mediated muscle atrophy – Publicly Invited Research 2016-2017
- A02 Shinohara
- A02 Maekawa
- A02 Ohgami
- A02 Nishimura
- A02 Kawano
- A02 Iwase
- A02 Furuichi
- A02 Myung
- A02 Kitamura
| Research Subject | Pathophysiological role of mitochondrial aconitase in unloading-mediated muscle atrophy |
|---|---|
| Research Group Leader |

|
| Research Collaborator(s) |
|
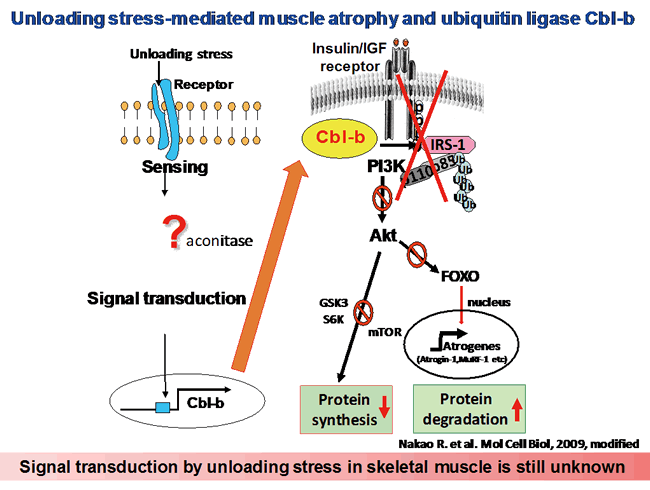
The impairment of growth factor signaling is a near-universal feature of skeletal myopathies induced by unloading. Clinical trials have established that during unloading, muscle tissue fails to respond to IGF-1, a dominant myotrophic hormone. Under normal conditions and in response to hypertrophic stimuli, IGF-1 promotes muscle growth and suppresses muscle loss largely through the Akt-dependent phosphorylation and cytosolic sequestration of FOXO transcription factors in skeletal myocytes, which leads to the inhibition of FOXO-dependent gene expression. In contrast, IGF-1-dependent Akt signaling is impaired during muscle atrophy, which decreases phosphorylation and increases transactivation of the FOXO target genes. In particular, FOXO regulates the expression of atrophy-related genes (atrogenes) that encode atrogin-1/MAFbx and MuRF-1, which are RING-type ubiquitin ligases and critical mediators of atrophic myopathies in vivo. Atrogin-1 and MuRF-1 regulate the degradation of key proteins involved in striated muscle growth and differentiation, including MyoD, calcineurin, and troponin-I. Although diminished growth factor responsiveness and enhanced proteolysis are both major atrophy-related processes, the mechanisms by which skeletal muscle becomes refractory to the trophic actions of muscle growth factors during unloading are not well defined.
We suggest that the ubiquitin ligase Cbl-b plays a major role in skeletal muscle atrophy induced by unloading. The mechanism of Cbl-b-induced muscle atrophy is unique, in that it does not apparently involve degradation of the structural components of muscle; instead, it impairs muscular trophic signals in response to unloading conditions. Recent studies of the molecular mechanisms of muscle atrophy have focused on the role of the IGF-1/PI3K/Akt-1 signaling cascade as a vital pathway in regulating the balance between hypertrophy and atrophy. These studies showed that under muscle-wasting conditions, such as disuse, diabetes and fasting, decreased IGF-1/PI3K/Akt-1 signaling augments the expression of atrogin-1, resulting in muscle atrophy. However, these studies did not address the mechanisms of the unloading-induced impairment of growth factor signaling. In the present study, we found that under both in vitro and in vivo experimental conditions, Cbl-b ubiquitinated and induced the specific degradation of IRS-1, a key intermediate of skeletal muscle growth regulated by IGF-1/insulin and growth hormone, resulting in the inactivation of Akt-1. The inactivation of Akt-1 led to the upregulation of atrogin-1 through the dephosphorylation (activation) of FOXO3, as well as reduced mitogen response in skeletal muscle. Thus, the activation of Cbl-b may be an important mechanism underlying the failure of atrophic muscle to respond to growth factor-based treatments such as IGF-1 (see figure below).
The Cbl-b gene is highly sensitive to mechanical stress (unloading). Therefore, we hypothesized that some components of the signal transduction pathways that regulate the expression of Cbl-b are important sensors of mechanical stress. We identified transcription factors that regulate Cbl-b expression using rat L6 myoblasts and differentiated myotubes (data not published). The elucidation of the biological relevance of Cbl-b expression as a sensor of unloading is a critical step in this project toward defining the molecular mechanism through which mechanical unloading is transduced into biochemical signaling in skeletal muscle.